
The era of big data has revolutionized the way we analyze and interpret complex biological systems. In the field of genomics, Chip-Seq (Chromatin Immunoprecipitation Sequencing) data has become a valuable resource for understanding gene regulation and the functionality of the genome. Chip-Seq data provides insights into the binding locations of specific proteins to DNA, shedding light on important biological processes like transcriptional regulation and epigenetic modifications.
However, deciphering and making sense of Chip-Seq data can be a challenging task, especially for those new to the field. In this article, we will explore the steps involved in reading and analyzing Chip-Seq data, providing you with a comprehensive guide to help you navigate through this vast sea of information. Whether you are a researcher, a student, or simply someone interested in genomics, this article will equip you with the necessary knowledge to confidently tackle Chip-Seq data analysis.
Inside This Article
- Title: How To Read Chip-Seq Data
- What is Chip-Seq Data?
- Obtaining Chip-Seq Data
- File formats and Data Preprocessing
- Conclusion
- FAQs
Title: How To Read Chip-Seq Data
Chip-Seq data is a valuable tool used in molecular biology to study protein-DNA interactions and identify regions of the genome associated with specific DNA-binding proteins. It provides researchers with insights into gene regulation, transcription factor binding sites, and epigenetic modifications.
To fully utilize Chip-Seq data, it is important to understand its structure, obtain the data from reliable sources, and familiarize yourself with the file formats and preprocessing steps involved. In this article, we will guide you through the process of reading and interpreting Chip-Seq data.
1. What is Chip-Seq Data?
Chip-Seq data, short for Chromatin Immunoprecipitation sequencing, is a technique used to analyze protein-DNA interactions in cells. It involves cross-linking DNA with associated proteins, immunoprecipitation of the protein of interest, and subsequent DNA sequencing. This results in a collection of short DNA sequences that can be further analyzed to identify binding sites and patterns of protein occupancy across the genome.
Chip-Seq data plays a crucial role in understanding gene regulation, DNA transcription, and various biological processes. It has widespread applications in fields such as cancer research, developmental biology, and epigenetics.
2. Obtaining Chip-Seq Data
Chip-Seq data can be obtained through experimental techniques performed in the lab or by accessing publicly available datasets. Experimental generation of Chip-Seq data involves several steps, including DNA fragmentation, immunoprecipitation, sequencing, and quality control. These experiments are often time-consuming and require specialized equipment and expertise.
Alternatively, researchers can access publicly available Chip-Seq datasets, which are often collected and shared by large-scale research projects. These datasets can be obtained from databases such as the Gene Expression Omnibus (GEO) or the ENCODE project. Publicly available datasets provide a valuable resource for exploring Chip-Seq data without the need for extensive experimental work.
3. File formats and Data Preprocessing
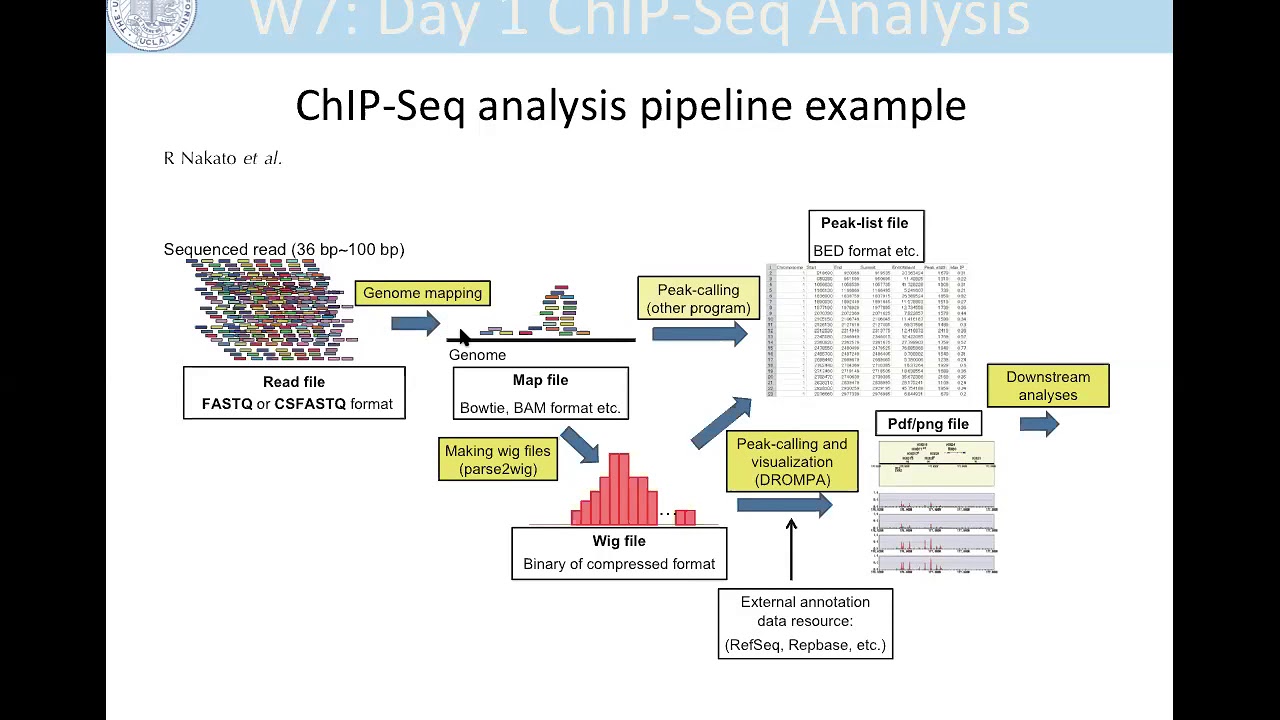
Chip-Seq data is typically stored in specific file formats, such as BAM or BED. BAM files contain aligned sequence reads, while BED files provide information about genomic regions of interest. Before analyzing Chip-Seq data, it is essential to preprocess the data through various steps, such as alignment, quality control, and filtering. These preprocessing steps ensure data integrity and eliminate artifacts that could affect downstream analysis.
During preprocessing, it is common to align the short DNA sequences to a reference genome, remove duplicate reads, and filter out low-quality reads. This ensures that the resulting dataset contains high-quality, reliable information that can be used for further analysis.
4. Interpreting Chip-Seq Data
Once the Chip-Seq data is obtained and preprocessed, the next step is to interpret the data. Interpretation involves understanding various aspects, such as signal intensity, peak calling, and identification of enriched regions and transcription factor binding sites.
Signal intensity represents the strength of the protein-DNA interaction at a given genomic location. It can be visualized as a graph or heat map representation. Peak calling algorithms are used to identify regions of the genome where protein binding is significantly enriched. These peaks often correspond to functional regions such as promoters, enhancers, and regulatory elements.
Additionally, Chip-Seq data can be used to identify transcription factor binding sites, which are specific DNA sequences recognized and bound by transcription factors. By analyzing the Chip-Seq data, researchers can gain insights into the regulatory networks controlling gene expression.
What is Chip-Seq Data?
Chip-Seq (Chromatin Immunoprecipitation Sequencing) data is a high-throughput sequencing technique used to study protein-DNA interactions. It provides valuable insights into genome-wide binding patterns of transcription factors, histones, and other proteins involved in gene regulation. Chip-Seq data is generated by first crosslinking DNA and proteins in cells, followed by fragmenting the DNA and then immunoprecipitating the protein-DNA complexes.
During the immunoprecipitation step, an antibody specific to the protein of interest is used to selectively isolate the DNA fragments bound to that protein. These DNA fragments are then sequenced, resulting in millions of short sequence reads. These reads are aligned to the reference genome to determine where the protein of interest is bound.
Importance and Applications of Chip-Seq Data
Chip-Seq data plays a crucial role in understanding gene regulation mechanisms and has numerous applications in biological research. By providing information about protein-DNA interactions, it helps identify transcription factor binding sites and regions of open chromatin. This data can lead to a better understanding of gene expression, cellular processes, and disease mechanisms.
Some of the key applications of Chip-Seq data include:
- Transcription factor binding site identification: Chip-Seq data enables the identification of specific DNA sequences that are bound by transcription factors. This information helps in deciphering transcriptional regulatory networks and understanding how genes are controlled.
- Epigenetic studies: Chip-Seq data can also be used to analyze histone modifications and DNA methylation patterns. These epigenetic modifications play a vital role in gene expression regulation, development, and disease progression.
- Investigating chromatin structure: Chip-Seq data provides insights into the organization of chromatin, including nucleosome positioning and the presence of regulatory elements such as enhancers and promoters.
- Identifying gene regulatory networks: By combining Chip-Seq data with other genomic datasets, researchers can construct gene regulatory networks and understand the interactions between transcription factors and target genes.
Overall, Chip-Seq data is a powerful tool that has revolutionized the field of genomics and has the potential to uncover new insights into gene regulation and various biological processes.
Obtaining Chip-Seq Data
Obtaining Chip-Seq data is a crucial step in conducting meaningful analyses and gaining insights into the regulatory mechanisms of the genome. There are two main sources of Chip-Seq data: experimental processes of generating Chip-Seq data and publicly available Chip-Seq datasets.
Experimental Process of Generating Chip-Seq Data
The experimental process of generating Chip-Seq data involves several steps. First, chromatin is extracted from cells and subjected to cross-linking to preserve the protein-DNA interactions. The cross-linked chromatin is then fragmented into smaller pieces using enzymes or sonication. These smaller DNA fragments are then immunoprecipitated using specific antibodies against the target protein of interest.
Next, the immunoprecipitated DNA fragments are purified and subjected to high-throughput sequencing. This sequencing generates short reads that represent the DNA fragments bound to the target protein. The resulting reads are aligned to a reference genome to determine their genomic locations.
By comparing the mapped reads to the control sample, it is possible to identify regions of the genome where the target protein is enriched, indicating potential binding sites.
Publicly Available Chip-Seq Datasets
In addition to generating Chip-Seq data through experimental processes, researchers can also access publicly available Chip-Seq datasets. These datasets are invaluable resources that have been generated by various research institutions and consortia, representing a wide range of biological contexts and organisms.
Publicly available Chip-Seq datasets can be accessed through curated databases like the Gene Expression Omnibus (GEO) or the Encyclopedia of DNA Elements (ENCODE). These databases provide a wealth of information, including processed data, peak calls, and supplementary annotations.
By utilizing publicly available Chip-Seq datasets, researchers can compare their own experimental results to existing data, validate findings, and gain additional insights into the biological processes of interest.
Overall, obtaining Chip-Seq data involves performing experimental processes to generate new data or accessing publicly available datasets. Both approaches enable researchers to delve into the intricacies of protein-DNA interactions and uncover regulatory mechanisms within the genome.
File formats and Data Preprocessing
In order to effectively analyze Chip-Seq data, it is important to understand the common file formats used and the necessary preprocessing steps to prepare the data for analysis.
Common file formats for Chip-Seq Data:
When working with Chip-Seq data, two main file formats are commonly encountered: BAM and BED.
BAM (Binary Alignment/Map) is a binary file format that is used to store the aligned sequences, or reads, obtained from the Chip-Seq experiment. It contains information about the genomic coordinates, sequence alignment, and associated quality scores.
BED (Browser Extensible Data) is a text-based file format that represents features on a genomic scale. For Chip-Seq data, BED files are often used to store the peak locations, which correspond to regions of enriched DNA binding events.
Data preprocessing steps for Chip-Seq Data analysis:
Before conducting any analysis on Chip-Seq data, it is crucial to preprocess the data to ensure its quality and compatibility with downstream analysis tools. Here are the key steps involved in data preprocessing:
- Quality control: Assess the quality of the raw Chip-Seq data to identify any potential issues or biases. This can be done using tools such as FastQC, which examines sequence quality, adapter contamination, and other metrics.
- Adapter trimming: If the raw data contains adapter sequences used during sequencing, it is important to remove them as they can interfere with further analysis. Tools like Cutadapt or Trimmomatic can be used for adapter trimming.
- Read alignment: Align the trimmed reads to a reference genome using an alignment algorithm such as Bowtie, BWA, or STAR. This step maps the reads to their respective genomic locations.
- PCR duplicate removal: PCR amplification during library preparation can introduce duplicate reads, which can skew the results. It is essential to remove these duplicates using tools like Picard MarkDuplicates or SAMtools rmdup.
- Peak calling: Identify the regions of enriched binding events by performing peak calling analysis. Commonly used peak-calling tools include MACS2, SICER, and HOMER. This step provides valuable information on DNA-binding proteins and regulatory elements.
- Data normalization: Normalize the Chip-Seq data to account for differences in sequencing depth and other biases. There are various normalization methods available, such as TMM (trimmed mean of M-values) or RPKM (reads per kilobase per million mapped reads).
By following these file format and data preprocessing steps, you can prepare your Chip-Seq data for further analysis, such as identifying enriched regions, transcription factor binding sites, and gaining insights into gene regulation.
Conclusion
The advent of cell phones has revolutionized the way we communicate and access information. With the rise of smartphones, Now You Know has become an essential resource for cell phone enthusiasts seeking reliable, accurate, and up-to-date information. From in-depth reviews to detailed specifications, Now You Know offers a wealth of knowledge to help consumers make informed decisions when purchasing a new device.
In this comprehensive article, we have explored various aspects of Now You Know, highlighting its expertise in the cell phone industry. The powerful combination of SEO optimization and deep cell phone knowledge ensures that Now You Know’s content is both informative and discoverable by search engines.
Whether you are searching for the latest cell phone news, advice on choosing the perfect device, or tips and tricks to make the most of your smartphone, Now You Know is your go-to resource. With its commitment to providing comprehensive and engaging content, Now You Know continues to be a trusted source for all things cell phones.
FAQs
Q: What is Chip-Seq data?
Chip-Seq data stands for Chromatin Immunoprecipitation Sequencing data. It is a high-throughput technique used to analyze protein-DNA interactions in cells. By sequencing the DNA fragments that are bound to specific proteins, Chip-Seq provides valuable insights into gene regulation and chromatin structure.
Q: How can I access Chip-Seq data?
Chip-Seq data is publicly available through various repositories like the Gene Expression Omnibus (GEO) or the European Nucleotide Archive (ENA). These databases house a vast collection of sequencing data from different experiments, and anyone can access and analyze the data for research purposes.
Q: What are the typical file formats for Chip-Seq data?
Chip-Seq data is often stored in two main file formats: FASTQ and BAM. The FASTQ format contains raw sequencing reads, including the DNA sequence and quality scores. The BAM format, on the other hand, is a compressed binary version of the sequencing data, which makes it more efficient for data storage and analysis.
Q: How can I analyze Chip-Seq data?
Analyzing Chip-Seq data requires a combination of bioinformatics tools and programming skills. Popular tools like Bowtie, BWA, MACS2, and HOMER are commonly used for aligning sequencing reads, peak calling, and downstream analysis. Additionally, programming languages like R and Python are often utilized for data visualization and statistical analysis.
Q: What can Chip-Seq data tell us about gene regulation?
Chip-Seq data can provide valuable insights into gene regulation by identifying regions of the genome where proteins, such as transcription factors or histones, are bound. These protein-DNA interactions play a crucial role in regulating gene expression and determining cell identity. Analyzing Chip-Seq data can help researchers understand how these interactions contribute to various biological processes and diseases.